 |
Experience is one thing you can't get for nothing. |
Home
Lab Resources
- Agarose Gel Electrophoresis
- Bacterial Streak Plate
- Bacterial Transformation
- DNA Ligation
- PCR
- Pipettors
- Plasmid DNA Isolation and Restriction Enzyme Digests
- Preparation of Agar Plates
Pre-lab Preparation
- Laboratory Citizenship & Performance
- BioSciences Lab Program Honor Code
- Molecular Biology Tips
- Writing Up Methodology
Additional Resources
Agarose Gel Electrophoresis of DNA
DNA is driven through the agarose matrix by electric current. Smaller or more compact molecules pass through the matrix easier and migrate farther than large molecules. All DNA has the same charge per unit length and linear pieces migrate according to size. The range of sizes separated in a gel is controlled by the % of agarose in the gel.
| Resolution Versus Matrix Concentration | |
|---|---|
Agarose |
Useful for Range of Linear dsDNA Molecules (kb) |
0.3 |
5 - 60 |
0.6 |
1 - 20 |
0.7 |
0.8 - 10 |
0.9 |
0.5 - 0.7 |
1.2 |
0.4 - 6 |
1.5 |
0.2 - 3 |
2.0 |
0.1 - 2 |
| *Information from Molecular Biology LabFax, ed. T. A. Brown, Academic Press, 1991 | |
| The mobility is proportional to the voltage applied at low voltage but increasing voltage decreases the resolution of larger fragments of DNA. A general guideline for agarose gels in 1xTBE is 5V/cm maximum for resolving fragment lengths greater than 2 kb. The distance between the electrodes serves as the length in the calculation. Higher voltages increase the temperature of the gel causing increased band width and distortion of the lanes. The agarose can also melt, especially the low melting point agarose sometimes used when DNA is to be recovered from the gel. The mobility is also influenced by the choice of buffer systems. Besides the Tris Borate EDTA, pH 8.3 (TBE) buffer used in our experiments, a Tris Acetate EDTA buffer (TAE) is preferred by some. The TAE buffer shifts the range of resolution toward higher fragment lengths. |  Ethidium |
The nucleic acids are visualized with ethidium bromide (EtBr). This fluorescent dye, which contains a tricyclic planar group, intercalates between stacked base pairs of nucleotides and, in this environment, fluoresces when excited with ultraviolet light; the fixed position of the planar group and its close proximity to the bases causes dye bound to DNA to display increased fluorescent yield compared to free dye.
- UV radiation at 254 nm is absorbed by DNA and transmitted to the dye, whereas radiation at 302 and 366 nm is absorbed by the bound dye
- energy is re-emitted at 590 nm in the red-orange region of the visible spectrum
NOTE: most commercial UV light sources emit at 302 nm, which yields slightly less fluorescence than at 254 nm but produces LESS nicking of DNA.
We will include EtBr in the gel only. In this case the dye extends the length of linear and relaxed circular DNA by about 15% (the molecules are more rigid which decreases their mobility). Supercoiled DNA is positively supercoiled by ethidium bromide. Thus, the mobility of supercoiled DNA with respect to linear and relaxed circular DNA varies with the concentration of ethidium bromide present during the run.If a DNA sample is too dilute to measure at 260 nm or is contaminated with other compounds that absorb in the UV range, the amount of DNA present can be estimated from the intensity of ethidium bromide fluorescence. Since the amount of DNA in a solution is proportional to the fluorescence emitted by ethidium bromide, the DNA quantity in an "unknown" solution can be estimated by comparing its level of fluorescence with the intensity of known amounts of DNA.
| The molecular weight DNA markers used in our study are Quick-Load® 1 kb DNA ladder (New England Biolabs, Catalog #N0468S). The ladder (see figure) produces eleven DNA fragments ranging in size from 500 to 10,000 base pairs (see picture). This ladder can be used to quantitate the amount of DNA in a sample; the mass of DNA in each band in the ladder has been calibrated. | 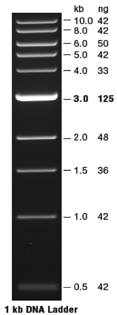 |
| The size of linear fragments of DNA is determined by comparison to standards: the log10 (# base pairs) is plotted versus distance migrated or Rf value {Helling R.B., Goodman H.M., and Boyer H.W. 1974. Analysis of endonuclease R-EcoRI fragments of DNA from lambdoid bacteriophages and other viruses by agarose-gel electrophoresis. J. Virol.14: 1235-1244}. | |
| The loading buffer (LB) combined with the DNA samples contains two tracking dyes, bromophenol blue and xylene cyanol, for visually monitoring electrophoresis and glycerol to make the sample dense enough sink to the bottom of the well; the stock solution is designed to be diluted about six fold in the sample. Bromophenol blue runs about the same size as a linear double-stranded DNA molecule of 300 base pairs in length in 1X TBE on a gel of 1% agarose. In low percentage gels of 0.4% agarose, the dye can emulate a 1000bp fragment. Remember not to run this dye off the bottom of the gel when you are trying to analyze small fragments. Xylene cyanol runs about the same as a linear double-stranded DNA molecule of 4kb in a 1% agarose gel. |
Agarose Gel Electrophoresis: Pouring and Running Gels
CAUTION
Agarose can become superheated and violently boil over. Exercise caution when heating. Swirl flask occasionally during heating. Heat until close inspection reveals that the agarose is 100% dissolved. Undissolved agarose will appear as little flecks that look like Lilliputian contact lenses.HEALTH HAZARD
Ethidium bromide (EtBr) is a powerful mutagen and a probable carcinogen; it's moderately toxic and should be handled with care. Wear gloves when handling contaminated equipment or solutions containing ethidium bromide. Confine the compound to the restricted area. After running the gels, use paper towels and plastic wrap to protect equipment and surfaces from being contaminated. Dispose of the gels in the Biohazard Waste Box. Do NOT put paper towels, plastic wrap, or gloves in this waste box--ONLY the gels.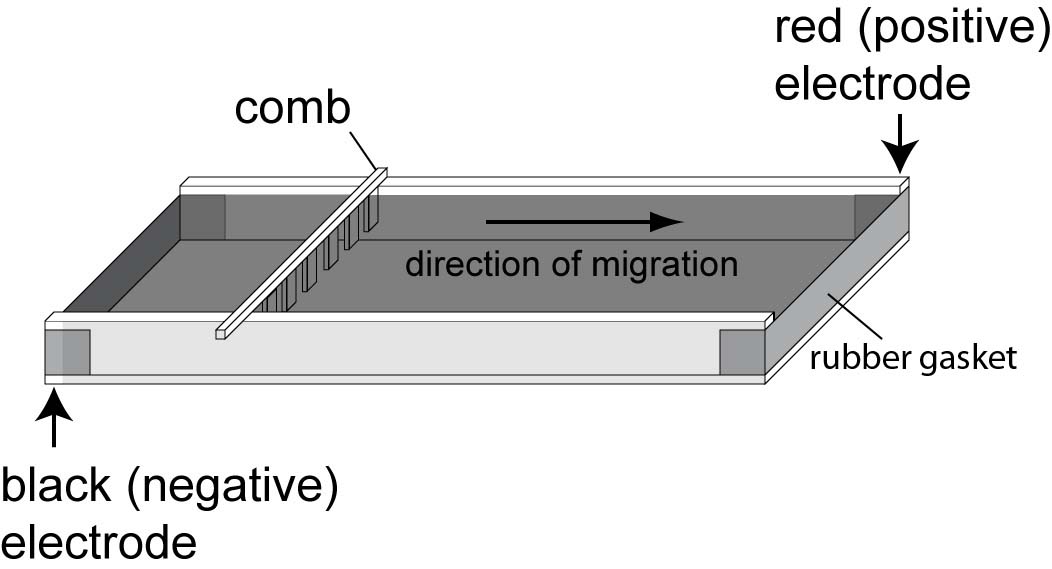
|
1. |
Prepare gel tray as diagrammed using an adjustable gel caster. Level the tray using a bubble level. |
|
|
|
2. |
Flasks of melted agarose (0.8 or 1%) in 1X
TBE have been prepared and held
at 50°C. Carefully pour melted agarose |
|
|
|
3. |
Let the instructor know when you are ready to add the
EtBr to your gel. After the instructor adds EtBr to
the gel, gently swirl the agarose. |
|
|
|
4. |
Immediately, pour the melted agarose into the level casting tray. Use a pipet tip to push bubbles towards the bottom of the gel. |
|
|
|
5. |
Let the tray cool until gel is translucent (takes at least 20 minutes). CLEAN UP ANY DRIPS ON THE BENCH AND RINSE THE BEAKER WITH RO-H2O! |
|
|
|
6. |
Prepare DNA samples. For agarose gels it is advisable to load the same volume into each well. Some samples may need to be diluted with water or TE to achieve this.
|
|
|
|
7. |
Turn lever on the caster to break the seal. Carefully remove comb and place casting tray into the electrophoresis unit for running. Fill unit with 1X TBE buffer to ~ 1 mm above gel. Pour buffer carefully onto the center of the gel to prevent the gel from sliding off the tray. |
|
|
|
8. |
Carefully load ALL of each sample into
different wells in the gel and record the order of the samples
in your notebook (record the location of each sample,
not just the ones that you loaded). Do
not press the tip into the bottom of the well while loading--allow
the sample to sink there. |
|
|
|
9. |
Position the lid and connect the electrodes so that
the anode is at the bottom of the gel (“run to
red”). |
|
|
|
10. |
Turn on the power supply (switch is located on the right side). Select “voltage” and
set the voltage to 130 V using the raise/lower arrow
keys; after the voltage is set, press the RIGHT SELECT button
until DISPLAY is lit to monitor the actual voltage (v) and
current (ma) during the run. |
|
|
|
11. |
STOP the run after ~ 45 min. Once the voltage is “zero” turn off the power supply, disconnect the electrodes, and carefully remove the lid. |
|
|
|
12. |
WEAR GLOVES. Drain excess buffer into the electrophoresis
unit and place casting tray with gel onto a paper towel
and carefully carry to the photography area. DO NOT
spread EtBr outside the designated area!! From this
point forward, assume that your gloves are contaminated with
EtBr--do not touch anything that is not supposed to be contaminated. |
|
|
|
13. |
Photograph the gel and compare the observed bands to the standards. The instructor or TA will take pictures for you using a UVP BioDoc-It™ System (components are listed below). |
|
Gel Documentation
ATTENTION: Avoid doing anything that would unintentionally contaminate the transilluminator or anything else with EtBr. For instance, do NOT lay gels directly on the transilluminator, but always on plastic wrap. Do NOT contaminate the equipment (knob, cover, table, etc.)--REMOVE your gloves FIRST.- Position the gel in the center of the UV transilluminator and smooth out any wrinkles in the plastic wrap
- With the cover in place, turn on the transilluminator
- Capture an image of the gel with your Smart phone or camera
Do NOT put paper towels, plastic wrap, or gloves in this waste box--ONLY the gels.
Estimation of DNA Amounts
Two methods are widely used to estimate DNA concentrations.Absorbance at 260 nm:At 260 nm, an absorbance (A) of 1 unit corresponds to a concentration of
- 50 μg/ml for dsDNA
- 40 μg/ml for RNA
- 33 μg/ml for ssDNA
- 20-30 µg/ml for oligonucleotides
Intensity of Ethidium Bromide Fluorescence: If a DNA sample is too dilute to measure at 260 nm or is contaminated with other compounds that absorb in the UV range, the amount of DNA can be estimated from the intensity of ethidium bromide fluorescence. Since the amount of DNA in a solution is proportional to the fluorescence emitted by ethidium bromide, the DNA quantity in an "unknown" solution can be estimated by comparing its level of fluorescence with the intensity of known amounts of DNA of similar size.
We would like to thank New England Biolabs for their generous support of our laboratory program
![]()
Visitors: to ensure that your message is not mistaken for SPAM, please include the acronym "Bios211" in the subject line of e-mail communications
Created by David R. Caprette (caprette@rice.edu), Rice University14 Jul 08
Author: Beth Beason Abmayr, Ph.D., Rice University
Updated 17 January 2015